2019-nCov, structurally speaking - part II
As a follow-up to my previous article 2019-nCoV, structurally speaking, I thought I’d share some of the news on the protein structure side and some insights from the structural analyses we’ve done at Discngine.
The first 2019-nCoV protein structure has been published
Wednesday this week, the structure of the 2019-nCoV main protease (Mpro) was finally released by the RCSB PDB. It’s an X-ray structure of Mpro in complex with a peptide-like inhibitor (See Figure 2), resolved by the Zihe Rao and Haitao Yang's research team at ShanghaiTech University.
The ligand, called inhibitor N3 by the authors, was first published in 2005 by Yang et al in a study on wide-spectrum anti-Corona virus drug design.
The new structure has been registered in 3decision. We took a look and thought I could share some of the findings here.

Figure 1. The peptide-like inhibitor N3
First, to check the quality of the resolved ligand structure, I loaded the electron density map (3decision retrieves the map directly from the PDBe server).
Despite the lack of electron density at the two extremes of the ligand, the ligand is well resolved and there is no ambiguity in the binding pose (See figure 2).

Figure 2. the newly resolved structure of the 2019-nCoV main protease in complex with a peptide-like inhibitor (PDB 6lu7).
N3 is a wide-spectrum Coronavirus Mpro inhibitor. Let's take a look at what the binding site looks like in a close homolog like SARS-nCoV. It turns out that there is only one residue that differs in the Mpro active site (see Figure 3).

Figure 3. The 3decision Sequence Viewer. To the left: Sequence alignment of the 2019-nCoV and SARS-nCoV orf1ab polyprotein sequences. Only the residues in the active site of the Mpro are shown. Differences compared to the reference sequence (here: 2019-nCoV) are shown by one-letter codes. Only one residue differs – S3309A. To the right: X-ray structure of the 2019-nCoV Mpro (6lu7) with the residue S3309 highlighted.
Before we finish, let’s drag in two homology models that were made before the Xray structure was released. As you can see in figure 4, the three structures are similar. As a reminder, Innophore’s homology model (in orange) has gone through an MD simulation and contains another ligand (Lopinavir). The Zhang Lab's model (in blue) comes directly out of the homology modeling.

Wide-spectrum virus protease structural comparison
The drug Lopinavir is known to bind to the main protease in HIV-1 and has been used by doctors in Thailand to treat 2019-nCoV infection. Several structures of the HIV-1-Lopinavir complex have been resolved and the a homology model of 2019-nCoV with Lopinavir bound is available. It would be interesting to compare the binding poses and active sites in HIV-1 versus 2019-nCoV.
The HIV-1 protease is a homodimer and the active site lies between two protein chains. This is not the case in the 2019-CoV binding site. In fact, the two binding sites are very different in terms of residue sequence. The structure overlay is therefore very tricky.
Luckily, 3decision automatically runs through a series of superposition methods to align structures. If the pocket sequences are too different in the two structures, 3decision will align using 3D-similarity parameters based on residue fingerprints. If that doesn’t work, 3decision will overlay the structure using a ligand superposition. In our example, the last method did the trick (See figure 5). The difference in protein fold can be seen very clearly. The ligands have a similar binding conformation however.
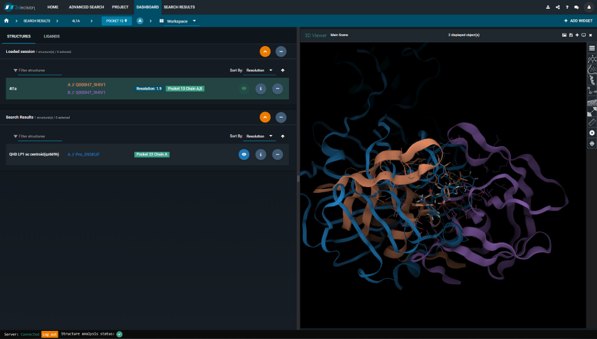
Figure 5. Overlay of the HIV-1 protease structure (PDB 4l1a) (protein chains colored in orange and purple) and the Innophore LP1 model of the 2019-nCoV protease (in blue). The overlay is based on the ligand-ligand superposition.
A new therapeutic target to combat 2019-nCoV?
BenevolentAI and the Imperial College in London are also contributing to global efforts to find a treatment for the viral outbreak. By searching within the BenevolentAI knowledge graph, they’ve identified a human kinase – the AP2-associated protein kinase 1 (AAK1) – as a potential target.
AAK1 promotes endocytosis, the process in which 2019-nCoV is believed to enter the host cell. An inhibition of AAK1 could therefore hinder the virus from infecting the cell. This hypothesis is supported by the fact that two AAK1-inhibitors (Sunitinib and Erlotinib) have been shown to stop dengue virus infection in mice.
The research team presents a high-affinity AAK1-inhibitor that they find to be best suited for the job: Baricitinib, a rheumatoid arthritis drug developed by Incyte and Eli Lilly (See Figure 6). According to activity data, Baricitinib has the advantage of binding both AAK1 and the Cyclin-G-associated kinase (GAK), another kinase involved in endocytosis. The researchers also argue that Baricitinib could be administrated in relatively low doses, compared to other AAK1-inhibitors, which would lower the risk for side effects.
To this date, I’m not aware of any planned trials with Baricitinib.
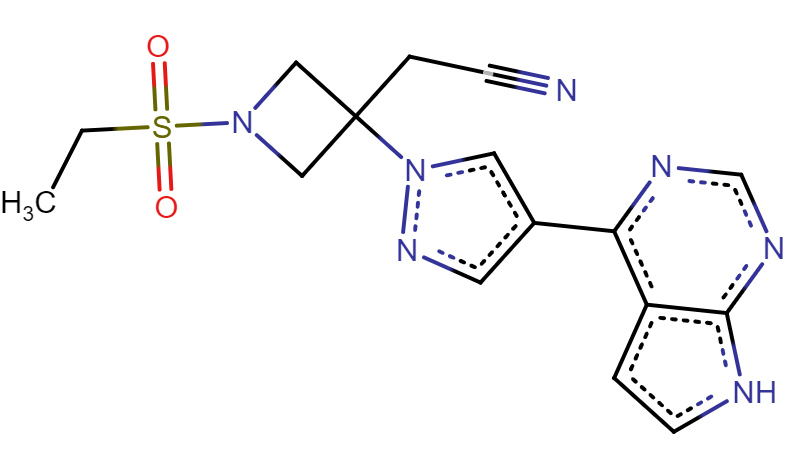
Figure 6. The drug Baricitinib
Now, let’s take a quick look at the structures. I found only one resolved protein structure with Baricitinib bound (PDB 4w9x). It’s a complex with the BMP-2-inducible protein kinase (BMP2K). By looking at the structure it’s clear that Baricitinib is a type-1 kinase inhibitor (See Figure 7). 3decision allows us to quickly overlay the BMP2K- Baricitinib complex with a resolved structure of AAK1. This gives us an idea of what the binding mode and the key interactions might look like in AAK1.

Figure 7. Structure overlay of BMP2K bound to Baricitinib (PDB 4w9x) in green, and AAK1 bound to an inhibitor in blue (PDB 5l4q). Both ligands are bound to the hinge.
It turns out that the BMP2K- and AAK1-binding-sites are extremely similar. By using the Sequence Viewer in 3decision, we can quickly verify where the sequence differences are situated, in 3D. We can also compare them to the Baricitinib-interactions (see Figure 8). In fact, only one residue-difference is in contact with the ligand: S63A. This “mutation” between the two Baricitinib-targets (i.e. BMP2K and AAK1) isn’t likely to influence the binding mode. The BMP2K-structure, therefore, gives a very good idea of what the AAK1- Baricitinib complex would look like.
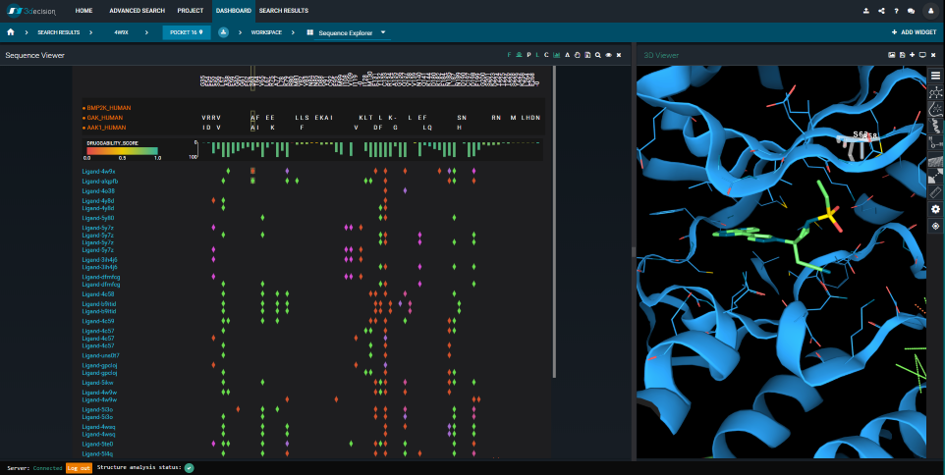
Figure 8. The 3decision Sequence Viewer. To the left: Sequence alignment of the BMP2K, AAK1 and GAK Mpro sequences. Differences compared to the reference sequence (here: BMP2K) are shown by one-letter codes. Ligand-protein interactions are mapped onto the sequence alignment and colored by type (green=hydrophobic, red=hydrogen bond, purple= water bridge, pink=salt bridge). Each line corresponds to a unique ligand. To the right: Structure overlay of BMP2K bound to Baricitinib (PDB 4w9x) in green, and AAK1 in blue (PDB 5l4q). The BMP2K protein chain, and the AAK1 ligand are hidden. BMP2K’s Ser63 is selected.
This is, in fact, a very nice demonstration of a scenario where you can answer "No" to the typical question “Do we really need to resolve a new structure do understand the ligand’s binding mode?”. Sometimes (more often than we think?) the structural data at hand is already sufficient.
To push this concept a bit further, my dear colleague Peter Schmidtke created a quick hybrid model of an AAK1-Baricitinib complex. He did this by grafting the ligand from PDB 4w9x into PDB 5l4q, and by launching a local minimization of the side chains around the new ligand. We've registered this model in 3decision for you to have a look (See Figure 9).

Figure 9. A model of an AAK1-Baricitinib complex.
What's accessible in 3decision?
Basically everything you see in this article!
We've registered the 2019-nCoV orf1ab polyprotein sequence available in GenBank. Since the sequence has not yet reached the UniProtKB, we are currently using a custom biomolecular identifier: Pro_093KUF. This is what you need to search for to find all 2019-nCoV Mpro structures in 3decision.
We've also created projects and session-links to simplify the navigation for you. I've centralized the direct access URLs here bellow. If you do not yet have access to 3decision and wish to contribute on this project, please send us an email on 2019-nCoV@discngine.com and we'll set up an account for you.